Abstract
Objectives: Congenital Lung Malformations (CLM) are rare and vary in their clinical presentations depending on the extent of lung involvement. Congenital Cystic Adenomatoid Malformation (CCAM), Congenital Lobar Emphysema (CLE) and Bronchogenic Cyst (BC) are all congenital lung malformations that present in imaging studies as abnormal air, fluid or fluid filled cysts. Neonates with CLM can present with respiratory distress at birth or may remain asymptomatic for a long period and are diagnosed incidentally on CXR in older children. Early diagnosis and prompt treatment in symptomatic CLM offers best chance for normal lung development. Thoracotomy or thoracoscopic lobectomy is the treatment of choice and leads to excellent long term results in children. This retrospective study presents single institution’s experience of common congenital lung malformations in neonates managed during the period from April 2013 till December 2014 with follow up of about 5 years.
Methods: This is a retrospective study of congenital lung malformations managed in our hospital from April 2013 to Dec 2014. Six patients were admitted in our NICU after birth due to respiratory distress and cyanosis. All of them needed mechanical ventilation due to acute respiratory failure. Three neonates were diagnosed as CCAM, two cases were diagnosed as CLE, and one neonate was diagnosed to have BC on CXR and confirmed on CT scan. Five neonates underwent lobectomy through thoracotomy and excision of BC was done in one case.
Results: Two neonates with CCAM were premature having gestation age of 32 and 34 weeks with birth weight of 1.9 Kg each. All other neonates were full term with birth weight ranging from 2.4 to 3 kg. The diagnosis was confirmed on histopathology. All the patients were on ventilatory support during immediate post operative period. The duration of NICU stay was from 1 to 3 weeks. The postoperative course was smooth in all the patients. All the neonates were cured and were discharged home in good health. All the children had a mean follow up of four years.
Conclusion: Congenital Cystic Adenomatoid Malformation (CCAM) is one of the important lung malformations treatable with surgery if managed properly. Congenital Lobar Emphysema (CLE) is life threatening but curable by timely surgery in neonates. Bronchogenic cysts (BC) are one of the congenital malformations that need simple excision. CCAM and BC may have malignant transformation in lator age in addition to other complications like infection and pneumothorax.
Introduction
Congenital Lung Malformations (CLM) constitutes a heterogeneous group of lung diseases [1-3]. Congenital Cystic Adenomatoid Malformation (CCAM), Congenital Lobar Emphysema (CLE) and Bronchogenic Cysts (BC) are three major congenital cystic lesions. They are uncommon but potentially life threatening [2,3]. They affects lower respiratory tract. Recently, there has been a significant increase in their diagnosis in the neonatal period because of use of CT scan, which is the gold standard for the diagnosis of CLM [3,4]. Patients with CLMs can present with respiratory symptoms at birth or they may remain asymptomatic for long period. When the diagnosis is delayed the children may present with chest infections and other complications [5]. The treatment of these lesions is surgical. CCAM, CLE and BC should be treated promptly in newborns for respiratory distress and pneumothorax [6]. Asymptomatic cysts in children should be removed in early age to avoid later complications which could make surgery more difficult [7]. Lobectomy in CCAM and CLE and simple excision in BC is the treatment of choice by thoracotomy or video assisted lobectomy. Because of potential complications of CLMs, surgical treatment should not be delayed [6-11]. This retrospective study presents single institution’s experience of common congenital lung malformations in neonates managed during the period from April 2013 till December 2014 with follow up of about 5 years.
Material and Methods
This is a retrospective study of Congenital Lung Malformations (CLM) managed in our hospital from April 2013 to Dec 2014. Six patients were admitted in our NICU after birth due to respiratory distress and cyanosis. All of them needed mechanical ventilation due to acute respiratory failure. Three neonates were diagnosed as Congenital Cystic Adenomatoid Malformation (CCAM), two cases were diagnosed as Congenital Lobar Emphysema (CLE), and one neonate was diagnosed to have Bronchogenic Cyst (BC) on CXR and confirmed on CT scan. Five neonates underwent lobectomy through thoracotomy whereas excision of BC was done in one case. Mechanical ventilation was continued postoperatively in all the six cases. All the neonates survived and were cured. They were discharged home in good health. In the mean follow up of four years, all the children are growing well and there is no difference in their respiratory functions.
Results
The mode of presentation, diagnosis on imaging and surgical treatment of all the neonates is summarized in the following (Table 1).

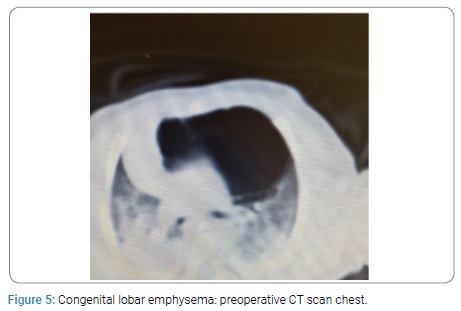
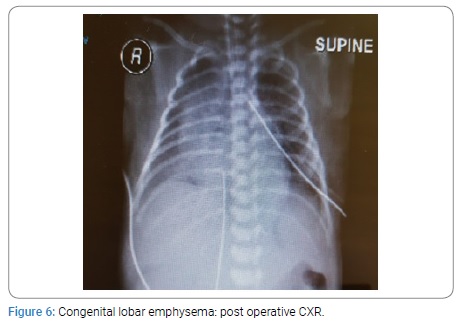
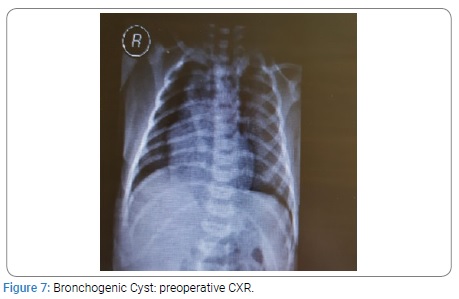
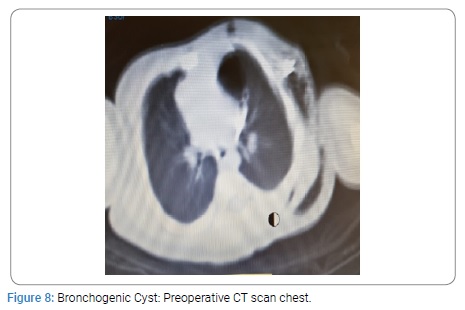
Two neonates with CCAM were premature having gestation age of 32 and 34 weeks with birth weight of 1.9 Kg each. All other neonates were full term with birth weight ranging from 2.4 to 3 kg. The diagnosis was confirmed on histopathology. All the patients were on ventilatory support during immediate post operative period. The duration of NICU stay was from 1 to 3 weeks. The postoperative course was smooth in all the patients. All the neonates were cured and were discharged home in good health. All the children had a mean follow up of four years. The post operative findings on CXR, CT scan and postoperative CXR of CCAM, CLE and BC are given below from (Figure 1-9).
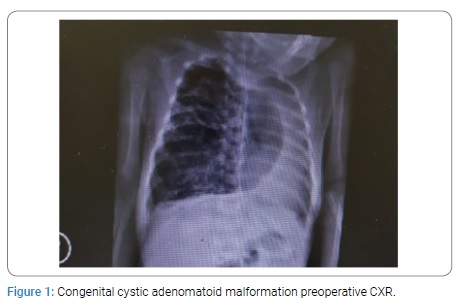
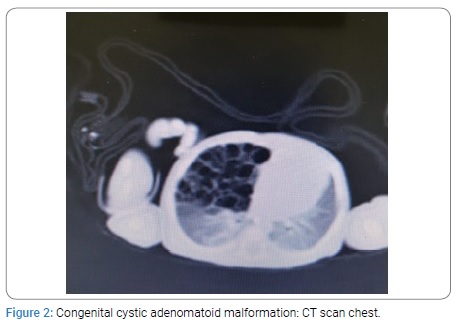
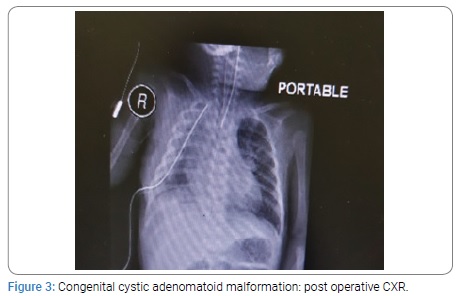
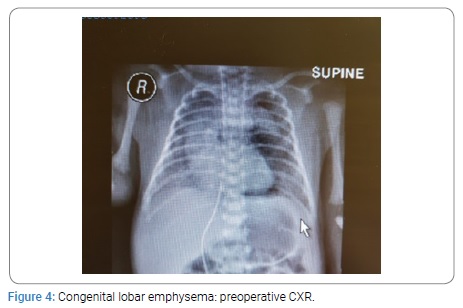
Discussion
Congenital Cystic Adenomatoid Malformation: The most common congenital lung malformations in neonates is Congenital Cystic Adenomatoid Malformation (CCAM) [1,8] Of all the cases 25% – 30% are CCAMs. However CCAM is a rare congenital malformation but potentially life threatening [4,8,9]. The incidence of CCAM is one case per 10,000 to 35,000 births [8]. Approximately 30% of patients of CCAM present with respiratory distress at birth which is its usual presentation [11-15]. Mortality is 25 to 30% in neonates. Cases with hydrops fetalis, poly-hydromnias and microcystic type have poor prognosis [12]. Prenatal ultrasound can diagnose CCAM antenataly [13,14]. In older children they present with history of recurrent pneumonia, respiratory difficulty, cyanosis and spontaneous pneumothorax with mediastinal shift [14]. CXR can show multiple cysts filled with air, fluid or air fluid. Ultrasound can detect CCAM but diagnostic errors can occur [7,16]. The differential diagnosis includes Congenital Lobar Emphysema (CLE), pulmonary sequestration, Bronchogenic Cyst (BC) and enteric cysts [16,17]. For diagnosis of CCAM, CT scan is mandatory. It determines the size of cysts and defines the extent of the lesion more accurately. Associated cardiac anomalies occur in 12 to 20% cases. Thus echocardiography has a role in patient’s evaluation. In symptomatic patients there is consensus that the lesion must be removed [18,19]. Lobectomy is the procedure of choice for the lesions confined to a single lobe and is the definite treatment. In general the surgical procedure can be safely performed in infants between 3 to 9 months of age. However some authors have recommended that surgery should be performed in neonatal period because neonates tolerate surgical stress better. Mean length of post-operative hospital stay is 2-3 days. Post-operative follow up evaluation of pulmonary functions generally show good results due to the fact that number of alveoli continue to increase till the age of 8 years. Thoracotomy and video assisted lobectomies are well tolerated by children as the rate of complications and deaths are low. Lobectomy in neonates and children is safe and easy to perform. It has been shown that CCAMs are hemartomatous lesions characterized by multi-cystic mass of lung tissue with proliferation of bronchial structure. The cysts are covered by columnar or cuboidal epithelium. There are reports of malignant transformations (carcinoma, pleuro-pulmonary blastoma) [1,7-9,11,19]. The management of asymptomatic lesions is controversial. However most authors recommend resection of CCAM because of risk of infection or neoplastic transformation later in life. The CCAMs vary widely in size and can affect part of a lobe, entire lobe or an entire lung. These lesions frequently affect lower lobes. However, any lobe or lung may be affected. The disease can be bilateral and may be associated with pulmonary hypoplasia. CCAMs are divided into 5 types from type 0 to type 4. Type 1 to type 3 are based on the size of cyst and histologic characteristics [8-24]. Type 0 has cyst smaller than 0.5 cm and is composed of tracheal or bronchial tissue, whereas type 4 consists of cysts larger than 5 cm and involves distal acinar tissue. In 1985, one group of authors classified CCAMs as macroscopic (larger than 5 cm) and microscopic (smaller than 5 mm).
Congenital lobar Emphysema: The second most common CLM is Congenital Lobar Emphysema (CLE). CLE is a rare lung malformation. A tertiary hospital may receive 1 to 2 cases per year. The incidence ranges from 1:20000 to 1:30000 births. It is twice as common in males as in females. The most common cause is deficiency of bronchial cartilage (25%) followed by bronchial stenosis/ bronchomalasia (25%). However in 50% of cases the cause remains unknown. CLE is characterized by hyperinflation of involved lobe of lung caused by air trapping resulting into distention. It leads to mass effect that compresses the remaining lung and causes mediastinal shift [1,2,4,10,12,20,21]. Most commonly CLE affects left upper lobe in 40 to 50% cases and middle lobe in 30 to 40% of cases whereas right upper lobe is involved in 20% of cases only. It may be bilateral in 20% of cases. Clinical presentation varies from asymptomatic to acute respiratory failure at birth. If the diagnosis is delayed then it may present with recurrent chest infections [18]. 0ne third cases of CLE are symptomatic at birth where as 50% are diagnosed in the first month of life. Majority of cases present with symptoms within six months of life [20-22]. Most patients present with moderate degree of respiratory dysfunction at birth, which worsens progressively as a result of hyperinflation [21-23]. This causes cyanosis which leads to respiratory failure. CLE is generally diagnosed clinically at birth on the basis of a profile of respiratory failure confirmed by CXR which shows hyperinflation of affected lung due to ball valve air trapping. There may be herniation of the lung across the midline, mediastinal shift and areas of atelectasis in the adjacent lung parenchyma. In some cases the affected lobe may appear opacified on CXR due to trapped amniotic fluid. CT scan plays an important role in confirming the diagnosis of CLE [20-23]. The differential diagnosis includes pneumatoceles, pneumothorax, pulmonary atelectasis and pulmonary hypoplasia. Histology is generally normal with alveolar dilation and increase in the number of alveoli in some cases. The treatment of choice in symptomatic patients is lobectomy through thoracotomy or video assisted lobectomy. The first case was operated in 1954 by Gross and Lewis. Pulmonary lobectomy is safe in children and results in a minimal loss of lung volume and there is good compensation due to lung growth during childhood up to 8 years of age. There is no long term difference in respiratory function [18,24]. Operative mortality is 3 to 7%. In the cases with conservative management the mortality increases from 50 to 70%. Thus CLE is life threatening condition but curable by timely surgery in neonates.
Bronchogenic Cyst: Bronchogenic Cysts (BC) are attached to tracheobronchial tree but without any communication with it. The lesions may be found along the treacheo bronchial tree, mostly in perihilar area and carina [17,25-32]. Usually they are single but multiple lesions may be found in some cases [17,27,28,30]. BCs are filled with fluid (mucous), air or air fluid. The natural course is uncertain. The common presentation of BC is respiratory distress due to compression of tracheobronchial tree, heart or both [29]. Some cases present with pneumo-mediastinum with surgical emphysema. Some BC remain asymptomatic in neonates and present with infection in later age. Although BC is benign but they may have malignant transformation if left untreated. BC are one of the congenital malformations which need simple excision [30-32].
Conclusion
Congenital Cystic Adenomatoid Malformation (CCAM) is one of the important lung malformations treatable with surgery if managed properly. Congenital Lobar Emphysema (CLE) is life threatening but curable by timely surgery in neonates. Bronchogenic Cysts (BC) are one of the congenital malformations that need simple excision. CCAM and BC may have malignant transformation in later age in addition to other complications like infection and pneumothorax.
Keywords
Neonates; Congenital lung malformation; Cystic adenomatoid malformation; Congenital lobar emphysema; Bronchogenic cyst; Lobectomy
Cite this article
Khan MH, Yaqub N and El Hatw M. K. M. Management of congenital lung malformations in neonates- northern area armed forces hospital experience. Clin Surg J. 2020;1(3):1–6.
Copyright
© 2020 Mumtaz H Khan. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
