Introduction
Subarachnoid hemorrhage continues to be a devastating disease for patients despite advances in treatment. The disease still portends a 40%–50% mortality rate, with patients who survive suffering from headaches, neurologic decline, and vasospasm. Furthermore, 70% of patients go on to develop vasospasm, and 30% of those patients become symptomatic in some manner. The largest risk associated with vasospasm is the development of delayed cerebral ischemia, which is a drop in the Glasgow Coma Scale score by two and is not attributable to other means. Suppose unrecognized and untreated can lead to long-term neurologic deficits and stroke. Emerging data indicate that this process is mediated by inflammation. The role microglia play in association with peripheral macrophages is ongoing. In this focused review, we highlight some of the emerging evidence that has linked inflammation to vasospasm and potential treatment modalities warranting further investigation (Figure).
Subarachnoid Hemorrhage and Vasospasm: The rupture of an intracranial aneurysm results in the sudden deposition of blood products in the subarachnoid space resulting in a complex cascade of physical and chemical disturbances [1]. The presence of subarachnoid blood causes an increase in Intracranial Pressure (ICP), which, correspondingly, results in a decrease in cerebral blood flow and oxygenation due to declines in Cerebral Perfusion Pressure (CPP) [2–4]. Increased ICP and blood in the ventricular system contribute to impaired cerebrospinal fluid absorption and circulation, causing the development of hydrocephalus and further increasing the intracranial pressure [2–4]. However, Subarachnoid Hemorrhage (SAH) may also result in delayed neurologic insults; after SAH, the resultant ischemia and metabolism of blood products activate cellular pathways that promote vasospasm [1,2,5,6].
Vasospasm refers to arterial vasoconstriction due to arterial spasm and may be either symptomatic or asymptomatic [1,6]. The incidence of vasospasm is highest on days 3 to 14 following aneurysm rupture but may persist to day 21 [6–8]. Radiographic evidence of vasospasm occurs in 30%–70% [9] of aneurysmal subarachnoid hemorrhage patients, but there is evidence that it may be as high as 90% [7,10]. When vasospasm results in regional hypoperfusion, it may result in clinically significant ischemia [11]. Vasospasm resulting in clinical deterioration is known as Delayed Cerebral Ischemia (DCI) [12]. Notably, DCI may result from multiple etiologies, but vasospasm is believed to be the most common [2,12]. DCI secondary to vasospasm is a significant cause of poor outcomes in patients with SAH [3,4,8] and occurs in approximately 20%–30% of patients following aneurysm rupture [9].
The management of vasospasm in subarachnoid hemorrhage involves monitoring for signs of vasoconstriction and neurologic deficits, administering prophylactic nimodipine, and intervening in delayed cerebral ischemia [3,13]. However, interventions for vasospasm are currently limited to endovascular administration of vasodilators [3,6].
The treatment of vasospasm in patients with aneurysmal subarachnoid hemorrhage is a significant challenge, likely due to its multifactorial etiology [3,5,6]. For example, studies of endothelin-antagonists have resulted in decreased arterial constriction without improvement in outcomes, and studies of the calcium channel blocker nimodipine have shown improved outcomes without a decrease in the incidence of radiographic vasospasm [5]. However, the lack of correlation between the direct treatment of the constricted vessels and neurologic outcomes in subarachnoid hemorrhage indicates a need for an improved understanding of the mechanisms of delayed cerebral ischemia.
Several identified mechanisms have been involved in the development of vasospasm and delayed cerebral ischemia. These include pathways for nitric oxide, endothelin, hypoxia-induced factors, and inflammatory markers (e.g., TNF, IL-1, IL-6) [1,14], and the interaction of these pathways and the role microglia play is an area in need of further investigation.
Microglia and Neuroinflammation: Inflammation is a process through which organisms respond to biological, physical, or chemical noxious stimuli that alter homeostasis [15]. Through various mediators, the inflammatory response attempts to remove the inflammatory stimulus, remove cells damaged from the inflammatory stimulus, and, ultimately, initiate tissue repair [15].
The anatomic structure of the Central Nervous System (CNS) is uniquely different from many peripheral organs as it lacks lymphatic vessels and has a Blood-Brain Barrier (BBB), which strictly regulates passage from the blood into the CNS [16]. Thus, leukocyte entry into the CNS occurs at lower levels than the periphery, and the CNS is known to possess a phenomenon known as an immune privilege [16–19]. Therefore, neuroinflammation is primarily mediated by microglia, which are resident macrophages of the CNS [17].
During homeostasis, microglia are in a “resting” state and function to maintain the stability of the CNS through surveilling the brain parenchyma for pathological changes and phagocytosing cellular debris, such as plaques and damaged or unnecessary synapses [16,17,20]. Once microglia encounter an inflammatory stimulus, they enter an “activated” state and trigger an inflammatory cascade, which attempts to—as previously described—remove the inflammatory stimulus, clear debris, and release signaling molecules to initiate tissue repair [15,16,21]. Regulation of this inflammatory response and initiation of tissue repair is paramount as prolonged inflammation may result in further damage [22]. Therefore, activated microglia may exhibit pro-inflammatory properties or anti-inflammatory properties; traditionally, these two phenotypes were described as M1 (classically activated macrophages) and M2 (alternatively activated macrophages) [21,23,24]. M1 microglia are pro-inflammatory and induced by interferon-γ (IFNγ), Tumour-Necrosis Factor (TNF), and Pathogen-Associated Molecular Patterns (PAMPs). In addition, M1 microglia produce greater amounts of Reactive Oxygen Species (ROS) and pro-inflammatory cytokines (IL-1, IL-6, IL-23), promoting greater microbicidal and tumoricidal capabilities [21,23,24]. However, to prevent indiscriminate damage to host tissues, M2 microglia mediate an anti-inflammatory response to resolve inflammation and promote tissue repair and remodeling [21,23,24]. M2 polarization is mediated by glucocorticoids, apoptotic cells, prostaglandins, immune complexes, cytokines (IL-4, IL-10, IL-13), and G-Protein Coupled Receptors (GPCRs) [21,23,24].
Although the traditional phenotypic classification of M1 and M2 is a useful paradigm in understanding the macrophage inflammatory response, additional studies have demonstrated that macrophage phenotypic expression is far more complicated, existing in diverse subspecialized states that overlap in gene expression and function [23–26]. Therefore, it is more appropriate to understand that macrophage activation exists as a continuum influenced by the complex milieu of environmental signals [23–26].
Microglia and Secondary Brain Injury: Primary brain injury occurs at the time of the initial insult, such as the mechanical forces imparted upon the brain during Traumatic Brain Injury (TBI). This initial insult triggers a molecular cascade that may result in additional, indirect damage. The injury caused by the sequela of the primary injury is a secondary brain injury. The secondary injury involves a complex interplay between inflammation, electrolyte imbalances, and alterations in Cerebral Blood Flow (CBF) [5,27,28].
The initial insult in SAH is the rupture of a blood vessel, most commonly a cerebral aneurysm, that results in the extrusion of blood under arterial pressure into the subarachnoid space [8,29]. This release of blood damages the cerebral parenchyma through physical stress [30], generation of free radicals [31], and the direct cytotoxic effects of hemoglobin [32]. Additionally, because the cranium is a rigid structure that enclosesis the brain parenchyma, blood, and Cerebral Sinus Fluid (CSF), the volume of these components is restricted within a fixed space. Therefore, an increase in the volume of these components must be accompanied by a commensurate decrease in another [27,33,34]. This demonstrates an inverse relationship between ICP and CPP: CPP = MAP-ICP [27,33,34]. After SAH, blood pooling in the subarachnoid space causes an increase in ICP and resultant cerebral ischemia [30].
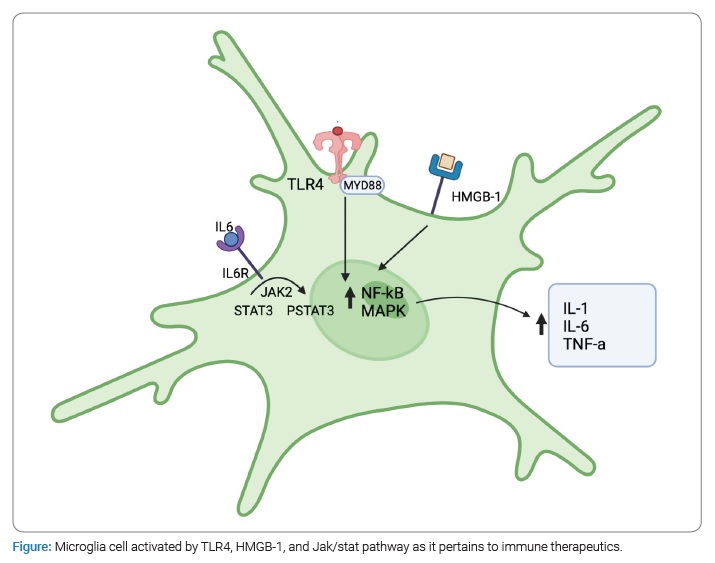
The noxious stimulus of SAH activates microglia, which significantly increase in number from 5 days to 8 days and peak between approximately 9 days to 15 days [30,35]. Expression of M1 markers—mediated by IL-6, TNF, and TLR4—predominates initially, increasing rapidly one day after SAH [36]. It is worth noting that heme has shown agonistic properties at TLR-4 [37]. Ultimately, this trend reverses on days 5 to 10 with the upregulation of IL-4 and TGF-β to promote an M2 phenotype [30,35]. Additionally, microglia appear to be the predominant mediator of inflammation after SAH for up to 7 days [38,39]. Evidence for this exists from an animal model, where the peripheral bone marrow was ablated and replaced with Green Fluorescent Protein-Tagged (GFP) leukocytes; there was no significant GFP infiltrate 7 days after SAH [39].
The predominance of an M1 is important for studying secondary brain injury. As mentioned above, TLR4 induces the polarization of the M1 phenotype and the continued release of pro-inflammatory mediators, like TNF. Furthermore, TLR4 and microglial appear essential in the pathogenesis of cerebral vasospasm through the production of TNF [40], as TNF induces Ras-related C3 botulinum toxin substrate 1 (Rac1), which mediates vasoconstriction [41]. Notably, TLR4 knockout mice possess resistance to cerebral vasospasm compared to wild-type mice [40]. Furthermore, the M1 response is accompanied by the release of ROS that directly damages the cerebral parenchyma and the further release of inflammatory cytokines, which breaks down the BBB and promotes edema [1,29,30,36]. This ensuing edema and leaky BBB increase ICP and decrease CPP, which may result in secondary ischemic injury [21,28,29]. Additionally, inflammatory products released by M1 microglia contribute to cerebral autoregulation dysfunction that creates a narrower cerebral blood flow (CBF) homeostasis window, which may result in ischemia or hyperemia [27].
Peripheral Immune Cross Talk: The presence of pyrexia, leukocytosis, and complement activation have previously suggested that the noxious stimulus of SAH incites systemic inflammation, despite the immunologic privilege of the CNS [42–46]. There is evidence that this systemic inflammation may facilitate the pathogenesis of post-hemorrhagic vasospasm, as additional studies have shown that fever, leukocytosis, and circulating immune complexes correlate with the development of cerebral vasospasm after SAH [42–45,47].
Direct evidence of the peripheral immune activation following SAH may be seen in the increased levels of cellular adhesion molecules after SAH [48]. Cellular adhesion molecules are endothelial transmembrane protein that stabilizes cell-cell interactions to facilitate leukocyte extravasation: the process through which leukocyte move from the circulatory system to the site of an inflammatory stimulus [16]. Leukocyte extravasation requires leukocytes to recognize an inflammatory stimulus, immobilize themselves against the sheer force of circulating blood, and pass through the endothelium; this process is commonly categorized as chemoattraction, rolling adhesion, tight adhesion, and diapedesis [16]. The unique structure of the BBB—particularly the presence of tight junctions—and low basal levels of cellular adhesion molecules limit leukocyte extravasation [16,49]. Notably, immune trafficking into the CNS appears to be limited to immunosurveillance by activated T cells, which appear to have less dependence on endothelial cellular adhesion molecule expression [50–52]. Therefore, the expression of cellular adhesion molecules is an important regulator of leukocyte extravasation, especially in the CNS [16]. In a rat animal model, intercellular adhesion molecule-1 (ICAM-1) was found to be up-regulated following SAH in a rat basilar artery model [53]. This has been corroborated by detecting elevated levels of three different cellular adhesion molecules after SAH: vascular cell adhesion molecule-1 (VCAM-1), ICAM-1, and E-selectin [48]. This evidence of peripheral immune activation was associated with a greater incidence of moderate or severe vasospasm in patient cohorts with the highest CSF levels of E-selectin (p = 0.044) [48].
Monoclonal antibody blocking experiments further provide evidence of the peripheral immune system and its implication in the development of vasospasm following SAH. In a rat femoral artery model, Oshiro et al. demonstrated that blocking the interaction between ICAM-1 and its integrins decreased the inflammatory response and vascular constriction [54]. Additionally, there was a reduction in the number of infiltrating macrophages and granulocytes in the periadventitial region of blood-exposed arteries (p = 0.0027) [54].
There is also evidence of a “crosstalk” between the peripheral immune system and microglial cells. Following SAH, microglia accumulation and pro-inflammatory gene expression are present and associated with degradation of the BBB, which would enable peripheral leukocyte extravasation [29,36–39,55]. There is also evidence that peripheral macrophages serve to modulate microglial function, suppress microglia-mediated phagocytosis, and reduce inflammation [56]. This suggests that the neuroinflammatory response may, in part, be concerted by this crosstalk.
Emerging Treatment Approaches: While the microglial inflammatory pathways activated after subarachnoid hemorrhage are complex and still poorly understood, some known traditional cell-signaling cascades are involved that allow the opportunity to theorize on potential therapeutic interventions that could mitigate inflammation and ultimately improve patient outcomes. The therapeutics we will discuss in this section are limited to those already used in practice today or currently undergoing clinical studies. As discussed previously, these therapeutics can involve targets ranging from common inflammatory cytokines and receptors all the way to intracellular signaling cascades [57].
Cytokines are well-understood inflammatory signaling molecules that play a role in a wide array of systemic inflammatory cascades [1]. IL-6 specifically is an inflammatory interleukin that has been previously shown to both have a strong correlation with worse outcomes after SAH and clinically shown to be upregulated in post SAH patient CSF; with a recent meta-analysis of 91 clinical and preclinical studies showing an increase of IL-6 in post-SAH patient CSF in 100% of patients included [1,58,59]. Tocilizumab, an IL-6 inhibitor, has been shown to correlate with decreased neuronal cell death, vasospasm, and micro clot formation when implemented in rabbit SAH models [60]. IL-1 is another inflammatory cytokine that has been shown to have increased levels in microglia 4 days–28 days after SAH [30]. In studies of aneurysmal SAH patients, IL-1 receptor antagonists (anakinra, canakinumab, rilonacept, etc.) administered twice daily from 3 days–21 days post ictus is shown to significantly reduce IL-6, CRP, and fibrinogen levels [61]. One clinical trial of IL-1 antagonist use in TBI showed a difference in mean microdialysate concentrations of macrophage-derived chemoattractant levels 45 times higher in a control group from that of the intervention group 5 days after administration of the antagonist (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4013762/). A third potential cytokine target is tumor necrosis factor-alpha (TNF-a), which is a target of medications such as adalimumab, etanercept, infliximab, golimumab, and certolizumab [62,63]. Current literature has shown enormous theoretical potential in the antagonism of TNF-a in SAH patients and is backed by SAH animal model studies showing that TNF-a inhibitors attenuate neuronal apoptosis, potentially via the Erk pathway [62,63]. Lastly, CCL2/ MCP-1, which is a cytokine best known for its role in regulating macrophage recruitment and polarization during inflammation, is thought to upregulate microglial HO-1 leading to their increased migration and proliferation during the inflammatory response after SAH [57]. Inhibition of CCL2, therefore, could dampen proinflammatory microglial response in the postictal setting. One such inhibitor, propagermanium, which targets glycoslyphopatidylinositol-anchored proteins closely associated with CCR and ultimately inhibits CCL2/ CCR2, has shown potential in the treatment of breast cancer patients [64]. Alternatively, bindarit is an oral CCL2 inhibitor and has been shown effective in suppressing the inflammatory responses of monocytes and macrophages in patients with diabetic periodontium [65].
C5a, a protein fragment released by C5-convertase, is another potential therapeutic target. While it is part of the complement cascade and known to be involved in the body’s innate immune response, its involvement in SAH remains poorly understood. Studies have shown that it is upregulated during SAH, and in studies of C5aR-/- mice with ICH, its knockout has been shown to significantly attenuate inflammatory response and subsequent neurological dysfunction (P = 0.017) [66]. Avacopan is an oral C5a receptor inhibitor, and IFX-1 is a monoclonal antibody against C5a that is both involved in clinical studies for ANCA vasculitis but could potentially offer benefit to SAH patients [67].
Toll-like receptors are integral membrane-bound receptors triggered by pathogen/ danger-associated molecular patterns (PAMPs/ DAMPs) that are vital to the innate and adaptive immune system. TLR4 signaling specifically has been demonstrated to be harmful in the early phase after a SAH, although the mechanism of which is poorly understood [68]. TLR4 antagonists, such as eritoran, ibudilast, JKB-121/122, NI-0101, and Cx-01, are a relatively nascent therapeutic class of drugs that are still undergoing clinical trial [69]. Unfortunately, trials investigating their ability to affect outcomes during non-SAH inflammatory processes have yielded poor results. For example, the ACCESS trial for eritoran showed nonsignificant mortality reduction in sepsis, and studies of ibudilast showed nominal reductions in CRP, TNF-alpha, and IL-10 levels (https://jamanetwork.com/journals/jama/fullarticle/1669798, https://onlinelibrary.wiley.com/doi/10.1111/adb.13182?af=R). Despite this, they may still present researchers with an opportunity to gain insight into the role of TLR4 in brain inflammation pathways and could potentially block some of its harmful sequelae in SAH patients.
Given the wide array of inflammatory cytokines and receptors involved in the post-SAH microglial response, it is no wonder that this process would encompass a number of both well and understood and some lesser understood cell signaling cascades [57]. One such target is the nuclear factor kappa-light chain enhancer of activated B cells (NF-kB), a well-studied protein complex that controls the transcription of DNA, cytokine production, and cell survival. Given the immense breadth of potential therapeutic coverage over NF-kB, with many of the therapeutics already discussed being involved in its signaling pathways, it would be beyond the scope of this paper to discuss every possible option that targets this cascade. Instead, our discussion will be limited to therapeutics with targets directly at Nf-kB transcriptional activity or targets along its related pathways (PI3K, Akt, MAPK, JAK/STAT) and already undergone clinical phase testing.
Pharmaceutical agents aimed at directly hindering NF-kB’s ability to induce transcription can be categorized into the following classes: selective inhibitors of nuclear export, which include slinexor, eltanexor,verdinexor, and felezonexor; histone deacetylase inhibitors which include vorinostat, romidepsin, belinostat, Panobinostat, tucidinostat; and DNA acetylation inhibitors like azacytidine and decitabine [70]. While many of these therapeutics are relatively new drugs undergoing phase 1, 2, or 3 clinical trials, they have shown promising results in treating various types of cancers and other chronic diseases. Furthermore, many established drugs across medicine are continually being discovered to have unknown additional NF-kB suppressing effects, leading to a rapid expansion of the list of potential therapeutic options in this pathway [71].
The PI3k/Akt pathway is hypothesized to play a protective role against SAH through inhibition of cell apoptosis and has led to investigations of numerous pharmaceutical up regulators of this pathway that could lead to antiapoptotic effects in post-SAH patients [72]. While the exact mechanism for the pathways’ antiapoptotic effect remains poorly understood, one of the leading theories is that it may promote M2 microglia phenotypes and, ultimately, resistance to oxidative stress [72]. Clinical research in PI3k/Akt activating therapeutics is currently being done with both novel pharmaceutical agents and with already clinically approved therapies newly found to upregulate this pathway. Although at the moment, the clinical trials of these agents are primarily in the realm of chemotherapeutics (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8005514/). Despite this, animal studies conducted with IGF-1 [73], R-568 [74], and alantolactone [75] have shown these agents capable of upregulation of PI3k/ Akt signaling and subsequent inhibition of neuronal apoptosis, inflammation, and polarization of microglia towards M2 phenotype. Thus, while the exact mechanisms of these agents are unknown, their potential for improvement of patient outcomes warrants continued study.
Mitogen-Activated Protein Kinase (MAPK) signaling pathway is upregulated during SAH and leads to the induction of pro-inflammatory cytokines, and its knockout in mice models of SAH is correlated with reducing both inflammatory cytokines and preventing microglial morphological activation [76]. Furthermore, animal studies into MAPK inhibitor SB203580 demonstrate a significant reduction in vasospasm, cellular apoptosis, and TNF-a levels compared to no intervention in a post-SAH setting [77]. Thus, there exists the potential to extrapolate and test these findings with other MAPK inhibiting chemotherapeutics (vemurafenib, dabrafenib, encorafenib, etc.) already being implemented in a clinical setting [78].
Janus kinase/signal transducers and activators of transcription (JAK/ STAT) signaling pathways are strongly associated with tumor growth and progression and play a role in cytokine secretion in many diseases [79]. The role of JAK/STAT in microglial activation during SAH has been well studied, and its inhibition in animal SAH models is shown to inhibit microglial conversion to the M1 phenotype and produce a reduction in neuronal damage and inflammatory protein levels [80]. Despite most of the clinical research on these agents pertaining to their use as chemotherapeutics, the accessibility of chemotherapeutic inhibitors of JAK/ STAT, both those that have been FDA approved (ruxolitinib, tofacitinib, etc.) and several others undergoing clinical trials, there exists an immense potential for further exploration of these therapeutic agents and their ability to modulate neuroinflammation in SAH patients.
A protein with potential as both a biomarker and therapeutic target for SAH treatment is the high mobility group box 1 (HMGB1). As a biomarker, it has been shown that CSF levels of SAH patients correlate with poor outcomes and can aid in interpreting neurological sequela [81]. In rat models of SAH, inhibition with anti-HMGB1 mAb leads to an attenuation of microglial activation and amelioration of neurological dysfunction [82]. It is believed that HMGB1 goes on to play a role in the inflammatory cascades that include RAGE, JAK/STAT, PI3K/Akt, NF-kB, and TLR4, many of which have already been discussed here for their potential as therapeutic targets during post-SAH inflammation. In addition, compounds such as resveratrol, glycyrrhizin, rhinacanthin, RIPK3, 4OGOMV, eNOS gene, and purpurogallin all have exhibited the potential to interact with HMGB1 and provide for further potential investigation in benefiting SAH patients [83].
